Действующий
115. (108) Для каждого цикла стерилизации должны быть оформлены записи с указанием времени полного завершения цикла, давления, температуры и влажности в камере во время процесса, а также концентрации и общего количества использованного газа. Давление и температуру необходимо регистрировать на протяжении всего цикла на диаграмме. Эти записи должны составлять часть досье на серию готовой продукции.
116. (109) Загрузку после стерилизации необходимо хранить под контролем в условиях вентиляции, чтобы обеспечить снижение содержания остаточного газа и продуктов реакции до установленного предела. Этот процесс должен пройти валидацию.
XX. Фильтрация лекарственных средств, которые не могут быть простерилизованы в окончательной упаковке
117. (110) Проведение стерилизующей фильтрации не является достаточным условием стерилизации, если возможно проведение стерилизации продукции в окончательной упаковке. Предпочтительным является метод стерилизации паром. Если продукция не может быть простерилизована в окончательной упаковке, то растворы или жидкости могут быть профильтрованы через стерильный фильтр с номинальным размером пор 0,22 мкм (или менее) или через фильтр с аналогичной способностью задерживать микроорганизмы в предварительно простерилизованные контейнеры (упаковки). Такие фильтры могут удалять большинство бактерий и плесневых грибов, но не все вирусы или микоплазмы. Поэтому должна быть рассмотрена возможность дополнения процесса фильтрации термической обработкой определенной степени.
118. (111) В связи с тем, что при стерилизующей фильтрации по сравнению с другими процессами стерилизации существует потенциальный дополнительный риск, непосредственно перед фасовкой рекомендуется повторная фильтрация через дополнительный стерилизующий фильтр, задерживающий микроорганизмы. Последнюю стерилизующую фильтрацию необходимо осуществлять как можно ближе к месту фасовки.
120. (113) Перед использованием стерилизующего фильтра и сразу после его использования необходимо проверять его целостность таким методом, как "точка пузырька", методом диффузионного потока или испытанием под давлением. При валидации должны определяться время, необходимое для фильтрации раствора заданного объема, и перепад давлений на фильтре. Любые существенные отклонения от указанных параметров во время текущего производства необходимо регистрировать и исследовать. Результаты этих проверок должны быть включены в досье на серию продукции. Сразу после использования необходимо подтверждать целостность критических газовых и воздушных фильтров. Целостность других фильтров должна подтверждаться через соответствующие интервалы времени.
121. (114) Не допускается использовать один и тот же фильтр в течение более одного рабочего дня, за исключением случаев, когда возможность более длительного его использования подтверждена валидацией.
122. (115) Фильтр не должен оказывать влияние на продукцию, задерживая ее ингредиенты или выделяя в нее какие-либо вещества.
123. (116) Частично укупоренные флаконы после завершения лиофильного высушивания должны находиться в зоне класса А до их окончательного укупоривания пробкой.
124. (117) Контейнеры (первичные упаковки) должны быть укупорены соответствующими способами, которые прошли валидацию. При использовании метода запайки, например, стеклянных или пластмассовых ампул, вся продукция подлежит 100%-ному контролю на целостность. В других случаях контроль целостности продукции должен проводиться установленными методами.
125. (118) Система укупоривания флаконов, наполненных в асептических условиях, не является полностью целостной до тех пор, пока на укупоренном пробкой флаконе не будет обжат (закатан) алюминиевый колпачок (крышка). В связи с этим обжим колпачка после укупоривания пробкой необходимо выполнять как можно раньше.
126. (119) Поскольку при обжиме колпачков может выделяться большое количество механических частиц, оборудование для обжима необходимо размещать отдельно и снабжать системой вытяжки воздуха.
127. (120) Обжим колпачков на флаконах может проводиться как часть асептического процесса с использованием простерилизованных колпачков или в условиях чистого помещения вне асептической зоны. В последнем случае флаконы должны быть защищены зоной класса А, пока не покинут асептическую зону, и в дальнейшем укупоренные пробками флаконы должны быть защищены путем подачи чистого воздуха класса А, пока на них не будут обжаты колпачки.
128. (121) Флаконы без пробки или со смещенной пробкой необходимо удалять до обжима колпачка. В случае если при обжиме колпачков необходимо вмешательство человека, для исключения прямого контакта с флаконами и минимизации микробной контаминации должна использоваться соответствующая технология.
129. (122) Эффективным средством защиты могут быть барьеры или изоляторы, ограничивающие доступ в рабочую зону, обеспечивающие требуемые условия и сводящие к минимуму прямой доступ человека к операции обжима.
130. (123) Первичные упаковки, герметизированные под вакуумом (вакуумные упаковки), должны проверяться на сохранение вакуума после заранее определенного промежутка времени.
131. (124) Первичные упаковки с продукцией для парентерального введения необходимо проверять индивидуально (поштучно) на наличие посторонних включений или других несоответствий по качеству. Визуальный контроль должен проводиться при установленных уровнях освещенности и фоне рабочего поля. Необходимо регулярно проверять зрение операторов, выполняющих визуальный контроль (если операторы используют очки, то проверка зрения проводится в очках). В ходе визуального контроля продукции рекомендуется достаточно часто организовывать перерывы в работе операторов. При использовании других методов контроля процесс контроля необходимо валидировать, состояние оборудования необходимо периодически проверять. Результаты визуального контроля должны быть оформлены документально.
132. (125) Испытание готовых продуктов на стерильность необходимо рассматривать только как завершающий этап в серии контрольных мероприятий, гарантирующих стерильность. Методика испытания на стерильность должна быть валидирована для каждого продукта.
133. (126) В случаях, когда получено разрешение на выпуск стерильной продукции по параметрам (Приложение N 17 к настоящим Правилам), особое внимание должно уделяться валидации и контролю всего технологического процесса.
134. (127) Выборка образцов продукции, которые были отобраны для проведения испытания на стерильность, должна быть репрезентативной для всей серии, и обязательно включать образцы, отобранные из тех частей серии, для которых предполагается наибольший риск контаминации, в частности:
а) (а) для продуктов, наполненных в асептических условиях, образцы должны включать контейнеры (первичные упаковки), в которые происходило наполнение в начале и в конце производства серии, а также после любого значительного вмешательства;
б) (b) для продуктов, прошедших термическую стерилизацию в окончательной упаковке, должно быть уделено внимание отбору проб из потенциально самых холодных частей загрузки.
** Справочно: Указания по проведению испытаний для подтверждения постоянного соответствия заданному классу чистоты приведены в стандарте ГОСТ Р ИСО 14644-2 (EN ISO 14644-2).
Производство биологических (в том числе иммунобиологических) фармацевтических субстанций и лекарственных препаратов
1. Технология производства биологических (в том числе иммунобиологических) фармацевтических субстанций и биологических лекарственных препаратов (далее - "биологические фармацевтические субстанции и лекарственные препараты") является критическим фактором, определяющим соответствующий регуляторный контроль. Фармацевтические субстанции и лекарственные препараты могут быть определены как биологические в значительной мере исходя из технологии их производства. Настоящее Приложение служит руководством для всего спектра биологических фармацевтических субстанций и биологических лекарственных препаратов.
2. Требования, установленные настоящим Приложением, распространяются на антибиотики на биологических стадиях их производства. Правила производства лекарственных препаратов, полученных из фракционированной донорской крови или плазмы, установлены Приложением N 14 к настоящим Правилам, а для нетрансгенных растительных лекарственных препаратов - Приложением N 7 к настоящим Правилам.
а) (а) Общее руководство (Часть А) - содержит дополнительные правила производства биологических фармацевтических субстанций и лекарственных препаратов, начиная от контроля посевных культур и банков клеток и заканчивая заключительными операциями и проведением испытаний;
б) (b) Специальное руководство по отдельным типам продукции (Часть В) - содержит дополнительное руководство для отдельных типов биологических фармацевтических субстанций и лекарственных препаратов.
а) (а) стадия производства: до того момента, пока биологические фармацевтические субстанции не станут стерильными, основным документом для них является глава IV настоящих Правил. Правила для последующих стадий производства биологических лекарственных препаратов содержатся в главе III настоящих Правил;
б) (b) вид продукции: настоящее Приложение является руководящим документом для всего спектра биологических лекарственных препаратов.
5. Эти два аспекта представлены в таблице N 1. Уровень требований усиливается по мере перехода от ранних к поздним стадиям производства биологических фармацевтических субстанций, однако принципы настоящих Правил должны соблюдаться всегда.
а) (а) для тканей и клеток, используемых для производства лекарственных средств, которые становятся биологическими фармацевтическими субстанциями для некоторых видов биологических лекарственных препаратов;
б) (b) в отношении высокотехнологичных лекарственных препаратов, в которых кровь или компоненты крови используются в качестве исходного сырья в части установления требований к отбору доноров, к качеству и безопасности при заборе, тестировании, обработке, хранении и транспортировке человеческой крови и ее компонентов;
в) (с) для производства и контроля генетически модифицированных организмов, в отношении которых должна быть обеспечена и соблюдена соответствующая изоляция и другие меры защиты на объектах, где осуществляется какая-либо работа с генетически модифицированными микроорганизмами, в целях установления и соблюдения соответствующего уровня биологической безопасности, - такие требования должны соблюдаться.
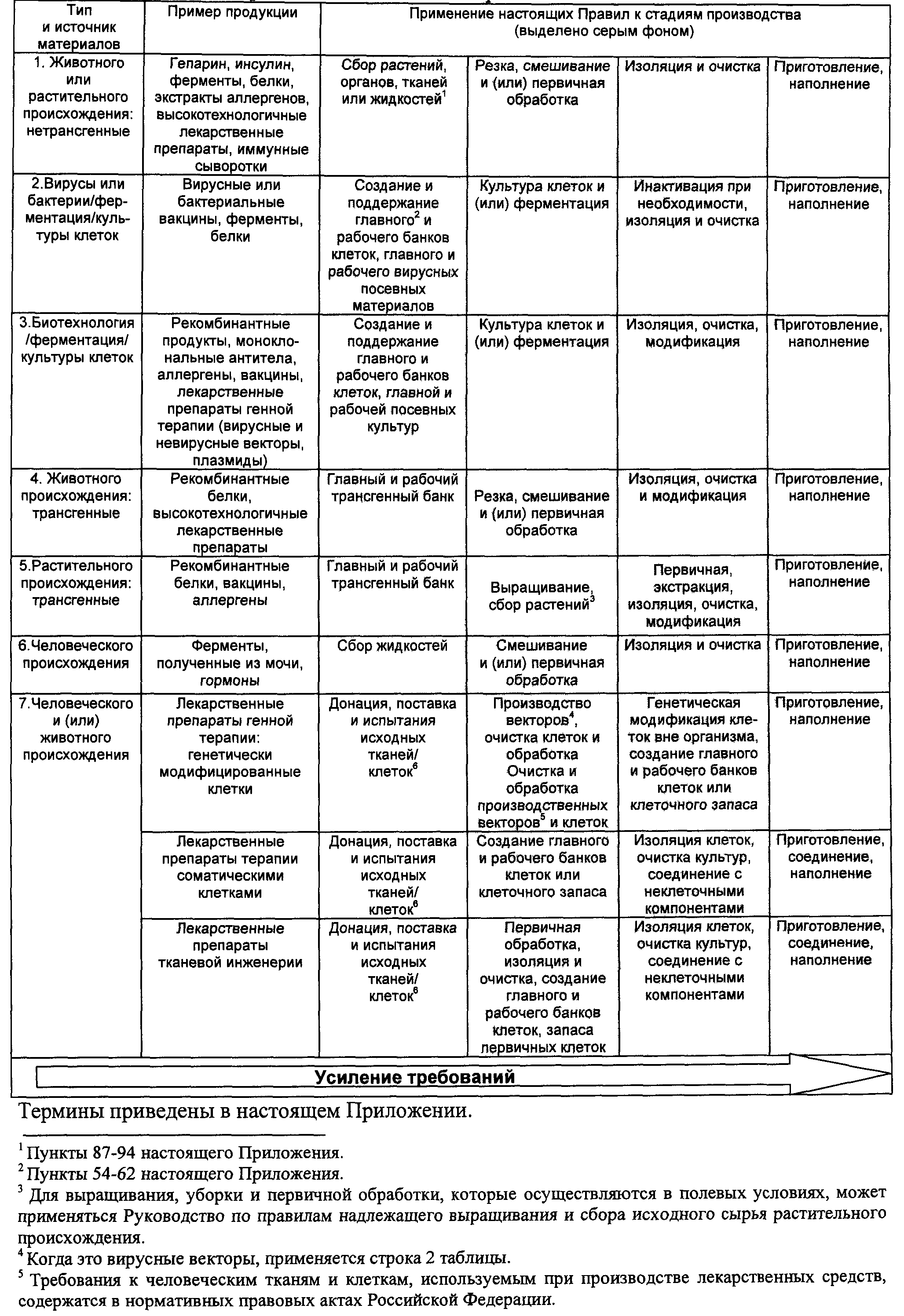
7. Производство биологических фармацевтических субстанций и лекарственных препаратов имеет свою специфику, определяемую характером продукции и технологией производства. Характер производства, контроля и применения биологических лекарственных препаратов требует особых мер предосторожности.
8. В отличие от обычных лекарственных средств, производимых с использованием химических и физических методов, способных демонстрировать высокую степень надежности, производство биологических фармацевтических субстанций и лекарственных препаратов включает в себя использование биологических процессов и материалов, таких как культивирование клеток или экстрагирование материала из живых организмов. Указанные биологические процессы могут демонстрировать свойственную им изменчивость, что приводит к существенному диапазону побочных продуктов различной природы. Поэтому принципы управления рисками для качества особенно важны для данного класса материалов и должны соблюдаться при разработке методов контроля на всех стадиях производства для минимизации вариабельности и уменьшения возможности контаминации и перекрестной контаминации.
9. Материалы, питательные среды и условия культивирования целевых микроорганизмов, культур клеток, вирусов во многом способствуют возможности роста контаминирующих агентов. Многие лекарственные препараты имеют ограниченную устойчивость к широкому спектру методов очистки, в особенности к методам, предназначенным для инактивации или устранения посторонних вирусных контаминантов. Для сведения к минимуму возможности такой контаминации производитель должен уделять основное внимание планированию технологического процесса, конструкции оборудования, помещений, систем обеспечения, условиям приготовления и добавления буферов и реагентов, отбору проб и обучению персонала.
10. Спецификации на продукцию (в частности, спецификации в общих фармакопейных статьях, фармакопейных статьях, регистрационное досье) должны определять, могут ли (и если могут, то до какой степени и до какой стадии) вещества и материалы иметь определенный уровень бионагрузки или же они должны быть стерильными. Производство также должно соответствовать другим требованиям, изложенным в регистрационном досье либо в протоколе клинического исследования (например, количество генераций (удвоений, пассажей) между посевной культурой или банком клеток).